New 3D method improves the study of proteins
27/04/2015
Current knowledge of the molecular bases of protein aggregation, the cause of several pathologies, has produced a series of algorithms to identify the regions of the proteins with a tendency to aggregate. Among them was AGGRESCAN, developed eight years ago by the same IBB researchers, which was one of the first computational methods created. However, the majority of these algorithms only analyse regions in the linear sequence of proteins. That makes it more difficult to predict the aggregation properties of globular proteins, where these sequences are often protected within their native spherical structure.
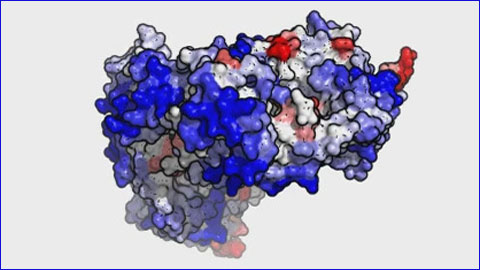
The AGGRESCAN3D (A3D), which was implemented as a web server freely accessible to the academic community, surpasses these limitations with an approach based on the protein structure in folded state. According to the article in which the researchers present the new method, published in Nucleic Acids Research, the new algorithm has a significantly higher precision than others based on linear sequences in predicting the properties of aggregation of globular proteins. It offers new and important features, such as the possibility to facilitate the modelling of pathogenic mutations and the design of proteins for therapeutic means, such as antibodies, with increases performance.
“Until now, A3D is the quickest algorithm available that can predict protein aggregation and work dynamically. In other words, it takes into account the flexibility of the protein structure. This allows us to model aggregations caused by natural fluctuations in the structure, as well as those caused by destabilising pathogenic mutations, and predict their impact on the protein's tendency to aggregate", states Salvador Ventura, IBB researcher from the UAB Department of Biochemistry and Molecular Biology and coordinator of the creation of the new method.
The new algorithm can be applied to any protein with a known structure or which can be generated by modelling. To validate the new method, researchers used proteins whose aggregation properties had already been characterised experimentally. In the static mode, it is possible to study individual proteins and protein complexes with up to 20,000 atoms and proteins with up to 400 amino acids in dynamic mode.
Protein Aggregation, a Key Issue in Biomedicine and Biotechnology
Protein aggregation has gone from being an area ignored in protein chemistry to becoming a key area of biomedicine and biotechnology.
“The incorrect folding of proteins and subsequent aggregation is the cause of a growing number of human disorders, such as Alzheimer's disease, Parkinson's and Type 2 Diabetes, and is one of the most important barriers in design and manufacturing of proteins for therapeutic means. These therapies, which imply the use of monoclonal antibodies, growth factors or enzyme replacement, have demonstrated to have an already high level of precision towards their molecular targets, and that is why continuing to study them is so significant”, Salvador Ventura concludes.
Article: Rafael Zambrano, Michal Jamroz, Agata Szczasiuk, Jordi Pujols, Sebastian Kmiecik and Salvador Ventura. AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Research, 2015 1. doi: 10.1093/nar/gkv359
Video: “Modelling of the monoclonal antibody RITUXIMAB used in the treatment of lymphoma and leukaemia. The areas in red correspond to the regions with greater propensity for aggregating, which could be redesigned with AGGRESCAN3D to improve the functionality of the drug.
How it Works:
http://biocomp.chem.uw.edu.pl/A3D/