

Disferlinopaties i antecedents familiars
Les distrofies musculars son un grup heterogeni de malalties musculars hereditàries, de les que se'n coneixen actualment dinou gens causants. El primer gen reconegut es el anomenat distrofina. Tots els pacients tenen debilitat muscular progressiva i canvis molt característics en la biòpsia muscular.
Les disferlinopaties constitueixen un grup d'aquestes distròfies musculars d'herència autosòmica recessiva amb mutacions en el gen de la disferlina (DYSF). Aquesta malaltia es pot presentar en diverses formes clíniques que inclouen: la miopatia de Miyoshi, amb un inici de la malaltia en els bessons, la distròfia de cintures de tipus 2B, en la que la malaltia s’inicia a la part proximal de les extremitats inferiors, i la miopatia d'inici a la part distal i anterior de les cames. Aquest gen codifica per a una proteïna anomenada disferlina que es localitza a la membrana de la fibra muscular i que s'ha involucrat en fenòmens de reparació de membrana i regeneració i diferenciació muscular. La disferlina també s'expressa en monòcits de sang perifèrica. la qual cosa va permetre al nostre grup desenvolupar un mètode no invasiu per avaluar els nivells d'expressió d'aquesta proteïna en els pacients sense necessitat de realitzar una biòpsia muscular.
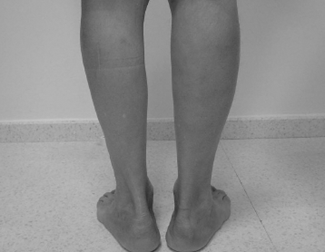
L'objectiu del nostre treball era descriure dos pacients amb una sola mutació al gen de la disferlina, és a dir en un al·lel, però que malgrat tractar-se d'una malaltia recessiva presentaven símptomes clínics. Aquesta conclusió es va poder efectuar, ja que teníem reconegudes les mutacions dels dos al·lels (canvi d'un aspàrtic per una tirosina: D625Y i canvi d'un glutàmic per una glicina E1734G) i una mutació homozigota (canvi d'una glicina per una arginina G519R) dels familiars afectes d'aquests dos pacients. A més de la debilitat muscular, els dos pacients presentaven nivells aixecats de CK i una RMN muscular anormal . La pacient 1 presentava infiltració grassa simètrica del tensor fasciae latae i atròfia asimètrica del adductor magnus. El pacient 2 mostrava àrees d'hiperintensitat en els gastrocnemious i tibialis anterior. La biopsia muscular mostrava canvis distròfics moderats i una expressió de disferlina reduïda a nivell de membrana muscular. El Western blot de monòcits i múscul esquelètic mostrava una reducció significativa de disferlina. Totes les altres proteïnes estudiades incloent caveolina-3 i calpaïna-3 presentaven un nivell d'expressió normal. La PCR a temps real mostrava uns nivells normals de ARN missatger de disferlina dels portadors respecte als seus parents afectes.
En resum, reportem per primera vegada que els portadors d'una única mutació en el gen de la disferlina poden ser simptomàtics. S'ha de pensar en aquest diagnòstic quan a més de debilitat muscular, CK aixecades i RMN muscular anormal, es troba una reducció (que no absència) de disferlina en múscul i/o monòcits. Aquest estudi amplia el llistat de patologies a pensar en front d'una debilitat muscular, encara que no hi hagi antecedents familiars. Afortunadament, l'examen inicial en monòcits fa que no sigui necessari fer una biòpsia muscular.
Referències
"Symptomatic dysferlin gene mutation carriers - Characterization of two cases". Illa, I; De Luna, N; Dominguez-Perles, R; Rojas-Garcia, R; Paradas, C; Palmer, J; Marquez, C; Gallano, P; Gallardo, E. NEUROLOGY, 68 (16): 1284-1289 APR 17 2007.
