

Disferlinopatías y antecedentes familiares
Las distrofias musculares son un grupo heterogéneo de enfermedades musculares hereditarias, de las que se conocen actualmente diecinueve genes causantes. El primer gen reconocido es el denominado distrofina. Todos los pacientes tienen debilidad muscular progresiva y cambios muy característicos en la biopsia muscular.
Las disferlinopatías constituyen un grupo de estas distrofias musculares de herencia autosómica recesiva con mutaciones en el gen de la disferlina (DYSF). Esta enfermedad se puede presentar de diversas formas clínicas que incluyen: la miopatía de Miyoshi con un inicio de la enfermedad en los gemelos, la distrofia de cinturas de tipo 2B, en la que la enfermedad se inicia en la parte proximal de las extremidades inferiores y la miopatía de inicio en la parte distal y anteriro de las piernas. Este gen codifica por una proteína llamada disferlina, que se localiza en la membrana de la fibra muscular y que se ha involucrado en fenómenos de reparación de membrana y regeneración y diferenciación muscular. La disferlina también se expresa en monocitos de sangre periférica, lo que permitió a nuestro grupo desarrollar un método no invasivo para evaluar los niveles de expresión de esta proteína en los pacientes sin necesidad de hacer una biopsia muscular.
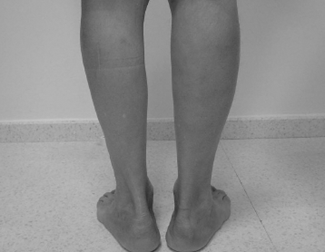
El objetivo de nuestro trabajo era describir dos pacientes con una sola mutación en el gen de la disferlina, es decir, en un alelo, pero que a pesar de tratarse de una enfermedad recesiva presentaban síntomas clínico. Esta conclusión se pudo efectuar, ya que teníamos reconocidas las mutaciones de los dos alelos (cambio de un aspártico por una tirosina: D625Y y cambio de un glutámico por una glicina E1734G) y una mutación homocigótica (cambio de una glicina por una arginina G519R) de los familiares afectos de estos dos pacientes. Además de la debilidad muscular, los dos pacientes presentaban niveles elevados de CK y una RMN muscular anormal. La paciente 1 presentaba infiltración grasa simétrica del tensor fasciae latae y atrofia asimétrica del adductor magnus . El paciente 2 mostraba áreas de híper intensidad en los gastrocnemious y tibilalis anterior. La biopsia muscular mostraba cambios distróficos moderados y una expresión de disferlina reducida a nivel de membrana muscular. El Western blot de monocitos y músculo-esquelético mostraba una reducción significativa de disferlina. Todas las otras proteínas estudiadas incluyendo caveolina-3 y calpaína-3 presentaban un nivel de expresión normal. La PCR a tiempo real mostraba unos niveles normales de ARN mensajero de disferlina de los portadores respecto a sus parientes afectos.
En resumen, reportamos por primera vez que los portadores de una única mutación en el gen de la disferlina pueden ser sintomáticos. Se ha de pensar en este diagnóstico cuando, además de debilidad muscular, CK elevadas y RMN muscular anormal, se encuentra una reducción (que no ausencia) de disferlina en músculo y/o monocitos. Este estudio amplía el listado de patologías en que pensar frente a una debilidad muscular, aunque no haya antecedentes familiares. Afortunadamente, el examen inicial en monocitos permite que no sea necesario hacer una biopsia muscular.
Referencias
"Symptomatic dysferlin gene mutation carriers - Characterization of two cases". Illa, I; De Luna, N; Dominguez-Perles, R; Rojas-Garcia, R; Paradas, C; Palmer, J; Marquez, C; Gallano, P; Gallardo, E. NEUROLOGY, 68 (16): 1284-1289 APR 17 2007.
