

ATM, la proteína que regula la reparación del ADN
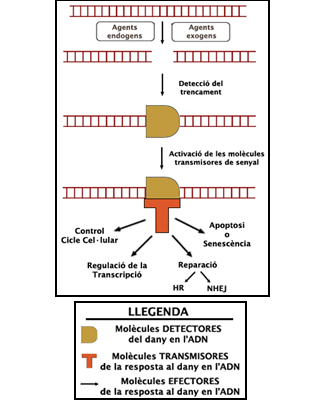
La presencia de lesiones o roturas en el ADN provoca una respuesta del sistema de Detección y Reparación del Daño (DDR) de la célula. Este sistema pone en marcha una activación en cascada de diferentes proteínas de reparación. ATM es una de estas proteínas y, es tan importante, que su ausencia provoca un síndrome humano denominado AT (ataxia-telangiectasia). Las células deficientes en ATM no reparan la totalidad del daño en el material genético, que se acumula durante bastante tiempo. Un equipo de científicos de la UAB ha investigado si la relación entre los bajos niveles de esta proteína y la persistencia de lesiones en el ADN puede deberse a una falta de reparación o a un problema en la detección de las roturas.
En los organismos eucariotas existen mecanismos de detección y respuesta en frente al daño en el ADN. Estos mecanismos se engloban en la DDR (DNA Damage Response): una cascada de activación de proteínas que se pone en marcha en respuesta a la presencia de daño y/o roturas en el ADN (Esquema 1, en la izquierda). ATM (Ataxia-telangiectasia mutated) es una proteína esencial en esta vía, donde realiza funciones de transmisión de la señal de daño. En las células deficientes en ATM la DDR no funciona correctamente, de forma que tienen problemas para reparar la totalidad del daño que les ha sido infringido: una proporción importante de las roturas del ADN (aproximadamente el 10-15%) permanecen sin reparar largo tiempo después de haber sido generadas (Kühne et. al., 2004), corriendo el peligro de que finalmente se dé una reparación incorrecta. La ausencia de ATM provoca una enfermedad denominada ataxia-telangiectasia (AT) en la que los pacientes presentan inmunodeficiencia y tendencia a sufrir linfomas y leucemias desde su juventud (Gatti et. al., 1988) (más información).
Dado que ATM es una proteína esencial en la transmisión de la señal del daño y que en su ausencia hay roturas del ADN que permanecen rotas, nos preguntamos si la deficiencia en ATM daba lugar a una carencia de reparación o bien a una carencia de detección de esta fracción de roturas. Para llevar a término este trabajo utilizamos células inmortalizadas provenientes de pacientes AT que fueron expuestas a dosis relativamente bajas de radiaciones ionizantes (1Gy). Tras la irradiación, las células se dejaron dividir una o dos veces y, pasado este tiempo, se localizaron las roturas no reparadas en metafases, donde el material genético está altamente condensado en forma de cromosomas y por lo tanto las roturas son visibles al microscopio.
Mediante immunofluorescència (detección fluorescente de proteínas) evaluamos la presencia de γH2AX en los cromosomas rotos. La histona H2AX forma parte del conjunto de proteínas sobre las que se empaqueta la doble hélice del ADN y es inmediatamente fosforilada -volviéndose γH2AX- cuando éste sufre un daño. La presencia de esta proteína es importantíssima para que otras proteínas de reparación sean reclutadas en el lugar del daño. Los resultados obtenidos demostraron que : (1) tal y como esperábamos, las células AT acumulan más roturas que las células normales y (2), aun cuando muchos de estos cromosomas dañados tienen γH2AX en el punto de rotura, una fracción importante -aproximadamente el 26%- (Figura 1 y 2) no están marcados con γH2AX.
Figura 1 y Figura 2.- En la izquierda, cromosomas rotos con y sin γH2AX; los cromosomas mitóticos presentan 4 telómeros (sus extremos naturales), 2 en la parte proximal y 2 en la parte distal y en la imagen han sido localizados y marcados en rojo. Cuando un cromosoma se rompe, sólo se observan 2 telómeros (*). En la derecha de cada imagen hay un dibujo esquemático de estos cromosomas rotos, sus telómeros y la presencia o ausencia de γH2AX (verde). En los cromosomas rotos de la izquierda (*) hay γH2AX en el lugar de la rotura, mientras que en el de la derecha (*) no lo hay. En la figura de la derecha, frecuencia de roturas en células AT y normales; las células AT acumulan más roturas que las células normales, y además, una de cada cuatro roturas NO presentan γH2AX (parte roja de la barra).
La ausencia de γH2AX en estas roturas sugiere que, probablemente, tampoco estén presentes otras proteínas de reparación. Para comprobar a ciencia cierta esta hipótesis se realizó una doble inmunofluorescencia de γH2AX y MRE11. MRE11 es una proteína detectora del daño en el ADN y una de las primeras al ser reclutadas en el lugar de la rotura. Por lo tanto, si la maquinaria funcionara correctamente, las roturas pendientes de ser reparadas habrían de estar marcadas con las dos proteínas utilizadas. Los resultados demostraron que, efectivamente, en ausencia de γH2AX tampoco hay reclutamiento de MRE11 en las roturas (Figura 3).
Figura 3.- Cromosomas rotos con y sin γH2AX y MRE11. Las cabezas de flecha indican cromátidas rotas. Las roturas de la izquierda tienen γH2AX (verde) y MRE11 (rojo), mientras que en la cromátida rota de la derecha no podemos localizar ninguna de las dos proteínas.
Estos resultados demuestran que las células AT acumulan más roturas que las células normales mucho tiempo después de haber sido irradiadas. Además, una cuarta parte de las roturas presentes en células AT no están siendo correctamente detectadas -debido a la ausencia de γH2AX y de MRE11- y aparecen como roturas invisibles para la DDR. muy probablemente esta invisibilidad favorece su persistencia en el tiempo e incrementa las posibilidades de que, más tarde, sean reparadas de manera incorrecta. Esta reparación incorrecta de roturas está muy relacionada con inestabilidad cromosómica y acumulación de mutaciones, procesos íntimamente implicados con el inicio de la carcinogénesis.
Financiación: Unión Europea (FI-CT-2003-508842); Fundació La Marató (TV32005-050110); Ministerio de Educación y Ciencia (SAF2004-20372-E, SAF2006-01653); Instituto de Salud Carlos III (RD06/0020/1020); Generalitat de Catalunya (2005SGR-00437).
Más información.Referencias
"Breaks Invisible to the DNA Damage Response Machinery Accumulate in ATM-Deficient Cells". Marta Martín, Mariona Terradas, George Iliakis, Laura Tusell, Anna Genescà. Genes Chromosomes Cancer, 2009, 48(9):745-59.
